
NaCl–KCl: The role of excess vibrational entropy
| dc.contributor | Materials Science and Engineering Laboratory, Ceramics Division National Institute of Standards and Technology, Gaithersburg, MD USA | en_US |
| dc.contributor.author | Burton, Benjamin P. | |
| dc.contributor.other | [email protected] | en_US |
| dc.date.accessioned | 2013-04-08T19:03:07Z | |
| dc.date.accessioned | 2014-08-05T19:24:54Z | |
| dc.date.available | 2013-04-08T19:03:07Z | |
| dc.date.available | 2014-08-05T19:24:54Z | |
| dc.date.issued | 2013-04-08 | |
| dc.identifier.citation | Chemical Geology Volume 225, Issues 3–4, 31 January 2006, Pages 222–229 | en_US |
| dc.identifier.uri | http://hdl.handle.net/11115/108 | |
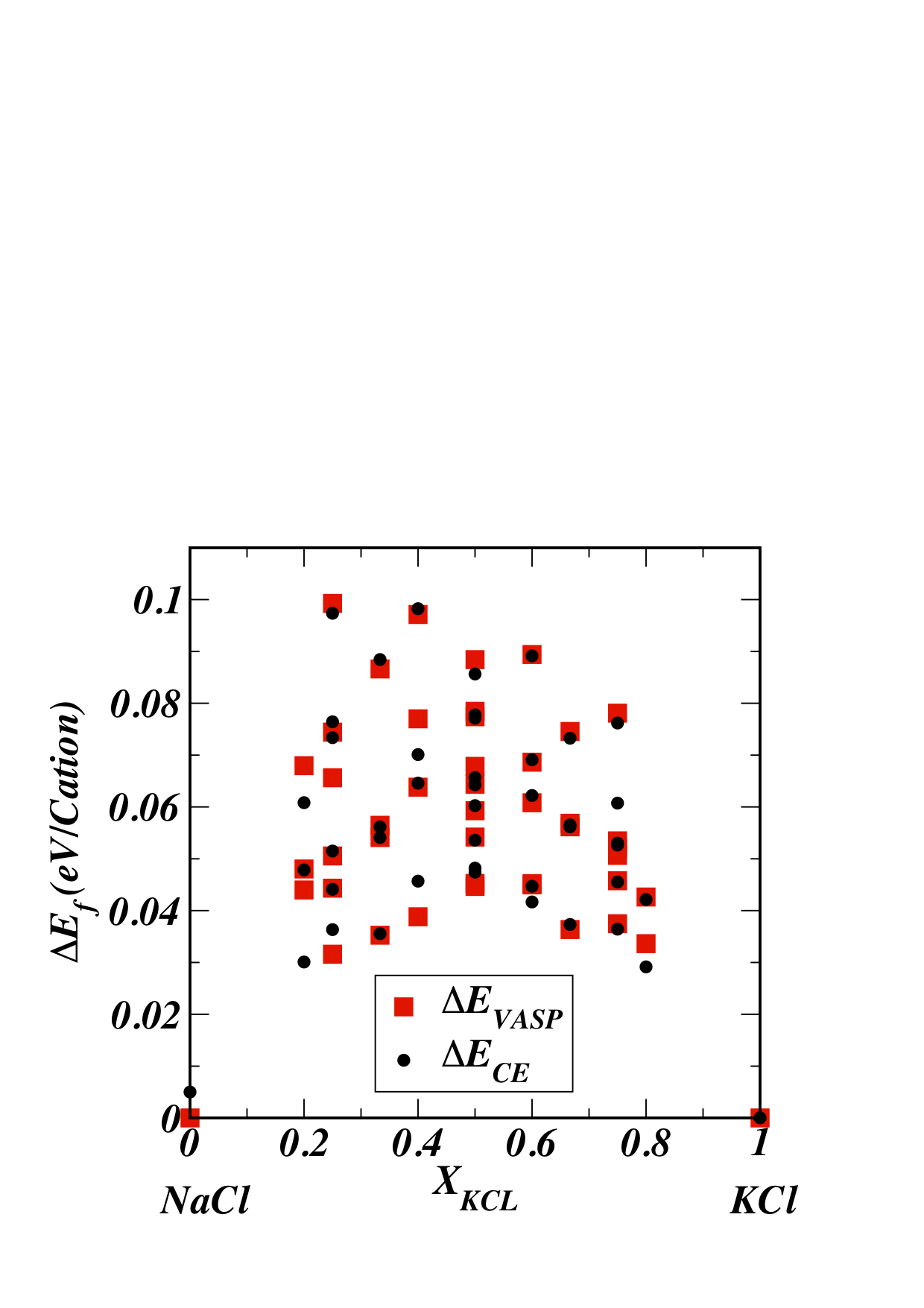
| dc.description.abstract | First principles phase diagram calculations were performed for the system NaCl–KCl. Plane-wave pseudopotential calculations of formation energies were used as a basis for fitting cluster expansion Hamiltonians, both with and without an approximation for the excess vibrational entropy (SVIB). Including SVIB dramatically improves the agreement between calculated and experimental phase diagrams: experimentally, the consolute point is {XC = 0.348, TC = 765 K}Exp; without SVIB, it is {XC = 0.46, TC ≈ 1630 K}Calc; with SVIB, it is {XC = 0.43, TC ≈ 930 K}Calc. | en_US |
| dc.relation.uri | http://dx.doi.org/10.1016/j.chemgeo.2005.08.016 | en_US |
| dc.rights | Attribution-NonCommercial-ShareAlike 3.0 United States | * |
| dc.rights.uri | http://creativecommons.org/licenses/by-nc-sa/3.0/us/ | * |
| dc.subject | Computational File Repository Categories::Methods::First Principles | en_US |
| dc.title | NaCl–KCl: The role of excess vibrational entropy | en_US |
| dc.type | Dataset | en_US |
Files in this item
{kind=link}
{kind=link}
This item appears in the following Collection(s)

Except where otherwise noted, this item's license is described as Attribution-NonCommercial-ShareAlike 3.0 United States